
Abstract
La1–xSrxCoO3 (x = 0, 0.1, 0.2) and LaCo0.8Fe0.2O3 perovskite nanoparticles were synthesized by spray-flame synthesis, and their electrocatalytic water oxidation activity was evaluated in the prepared state. Highly crystalline, rhombohedrally distorted cubic structures of the cobaltite perovskites were confirmed by STEM and XRD analyses. The decreased JT distortions were observed in the cobaltite perovskite structure upon substitution with Sr, while orthorhombic distortions in Fe-substituted perovskites could explain the changes in their Raman spectra. Debye–Waller factors and coordination numbers from fitted EXAFS data indicate more disordered crystalline structures upon Sr substitution and a lower Co–O coordination number at 20 at% Sr. Thermal characterization of the catalysts by STA coupled with QMS shows higher mass losses in Sr-substituted catalysts, and these results were associated with a higher concentration of carbonate species in these catalysts, which was also confirmed by XPS measurements. Both Fe and Sr substitution lead to higher catalytic OER activity of the cobaltite perovskites with lower overpotentials of about 30–50 mV. The cobaltite perovskite catalyst substituted with 20 at% Sr exhibited the highest OER activity and stable electrocatalytic performance at moderate conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Generation of hydrogen with renewable electricity is a key technology toward renewable and sustainable energy production. For this purpose, alkaline (AEC), proton-exchange membrane (PEMEC) electrochemical cells have been developed for water electrolysis [1, 2]. In these systems, the anodic oxygen-evolution reaction (OER) is the bottleneck reaction due to the involvement of a four-electron transfer process and a high number of intermediate species of surface adsorbates, which increases the overpotential requirement and thus, the cost of the electrolysis process [3, 4]. Additionally, the high cost and scarcity of the benchmark catalysts IrO2 and RuO2 has triggered investigations of transition metal-based oxide catalysts to develop earth-abundant, cheap, OER-active and stable electrocatalysts [5, 6]. Materials selection for these oxide catalysts are based on critical OER descriptors some of which include the bonding strength of metal hydroxide/oxyhydroxide [7], the concentration of oxygen vacancies [8], the number of 3d electrons at the surface transition metals with eg symmetry [9], as well as the elastic strain [10] and surface metal–oxygen coordination [11]. More recently, different kinds of machine learning strategies have also been developed to predict OER descriptors and suitable catalysts for water oxidation catalysts [12, 13].
Transition metal-based perovskite oxides (i.e., ABO3 compositions with A being rare-earth ions and B being transition-metal ions) are promising materials as water oxidation catalysts owing to their high ionic and electronic conductivity, structural stability, and chemical versatility [14]. A recent report on intrinsic OER activity of different perovskite oxides shows that the semiconducting LaCoO3 is one of the most active catalysts, having more than one order of magnitude higher OER activity compared to metallic LaNiO3 [15]. The report emphasizes that near-Fermi-level d-orbitals in cobaltite perovskites have an important effect on enhancing electron conduction and OER catalysis. The spin-state configuration of trivalent cobalt ions in LaCoO3 determines the degree of eg filling, and Co3+ ions in intermediate spin states show the highest OER activities as they lead to higher charge transfer rates [14]. Substitutional doping of A- and B-site cations in LaCoO3-type structures is also widely used to tailor their electronic properties for OER [16]. For instance, partial substitution of La3+ ions by Sr2+ ions in La1–xSrxCoO3–δ perovskites induces a notable increase in their OER activities with increasing Sr concentrations. These perovskites show OER activities comparable to those of IrO2 benchmark catalysts due to increasing bond covalency of Co–O, and high diffusivity of oxygen ions with Sr substitution [17]. On the other hand, the substitution of Co with Fe or Cr increases the overall density of Co 3d states near the Fermi-level, thereby enhancing the charge transfer between transition-metal ions and adsorbates during OER catalysis [15]. A low concentration of these B-site substitutes is considered beneficial to improve the OER activity of LaCoO3, while not drastically reducing the perovskite’s electronic conductivity. Similarly, A-site substitution at low concentrations is beneficial in terms of keeping structural stability of LaCoO3 during OER and reducing surface segregation of A-site cations [18]. Thus, it is of interest to investigate 0–20 at% A- and B-site substituted LaCoO3 as promising catalysts for OER.
Pristine and substituted perovskite cobaltites have been prepared as electrocatalysts for water oxidation by numerous techniques. Most recently, 3d inverse opal non-stoichiometric LaCoO3–δ was prepared by colloidal template techniques [19], and Pr0.5Ba0.5CoO3–δ perovskites were deposited on NdGaO3 substrates using pulse-layer deposition [20]. Also, LaCo0.7Mn0.3O3 particles were previously prepared by hydrothermal synthesis [21], while LaCo1–xFexO3 and La1–xSrxCoO3 particles were synthesized by sol–gel and molten-salt synthesis processes [22, 23]. Lately, gas-phase synthesized perovskite cobaltites have been shown as promising OER catalysts in our reports [24, 25]. Lab-scale spray-flame synthesis enabled the production of nanosized LaCoO3 and LaCo1–xFexO3 particles with enhanced OER activities compared to the other Co-based catalysts in the literature [26]. Owing to its high temperature and low residence time, spray-flame synthesis enables a scalable synthesis of crystalline, high surface-area nanoparticles. The gas-phase nucleation process provides an ideal mixing of dopant metal ions in perovskite nanoparticles and allows the synthesis of various cation-substituted perovskite cobaltites. Spray-flame synthesized La1–xSrxCoO3 perovskites have not yet been studied as OER catalysts. However, considerable amounts of unburned organic combustion residuals are typically adsorbed on the as-prepared high surface-area perovskites, and mild annealing at ~ 250 ºC is necessary to clean them from these carbonaceous residues. For the direct use of the catalysts, it would be advantageous if such a post-annealing can be avoided.
In the present work, we report the preparation of pristine, Fe- and Sr-substituted LaCoO3 nanoparticles as OER catalysts in a single step through spray-flame synthesis. The influence of cation substitution on the crystallographic and electronic structure as well as the surface composition of LaCoO3 perovskites was elucidated through structural, spectroscopic and thermal characterization techniques. The perovskite substitutions that led to the highest OER activity were determined.
Experimental section
Materials
The metal precursors of lanthanum(III) acetate hydrate (La(CH3COO)3·xH2O, 99.9%, Sigma-Aldrich), cobalt(II) acetate tetrahydrate (Co(CH3COO)2·4H2O, > 98%, Sigma-Aldrich), strontium(II) acetate (Sr(CO2CH3)2, 99%, Sigma-Aldrich), and Fe(II) acetate (Fe(CO2CH3)2, > 99%, Sigma-Aldrich) were used to synthesize Sr- and Fe-substituted LaCoO3 nanoparticles. Propionic acid (> 99%, Sigma-Aldrich) and 1-propanol (anhydrous, 99.7%, Sigma-Aldrich) were used as solvents. TGA measurement was used to calculate the total amount of crystallization water in lanthanum(III) acetate hydrate, and La(CH3COO)3·1.13H2O was determined as its chemical formula.
Synthesis of catalysts
A homemade spray-flame reactor allowing full control of gas flows and pressure was used to synthesize perovskite nanoparticles. The reactor setup was illustrated in our previous reports [26,27,28,29], and a representative photographic image of the spray flame observed during the synthesis of LaCoO3 is shown in Fig. S1 in the supplemental material. For the preparation of the precursor solutions, a mixture of 1-propanol and propionic acid was initially stirred in a glass flask for 5 min at room temperature. The total metal concentration of La-Sr-Co and La-Co-Fe was adjusted to 0.2 M, and the La-, Co-, Sr- and Fe-acetate-based precursors were then added to the solvent mixture. The solution in the flask was set inside a thermalized silicon oil bath (60 °C). The precursor salts were then stirred for at least 1 h until all precursors were fully dissolved. The nominal atomic ratios of Sr/(Sr + La) were selected as 0, 0.1, and 0.2, while Fe/(Fe + Co) ratio was adjusted to 0.2. Table S1 summarizes the precursor solution characteristics used for the syntheses.
For the spray-flame synthesis process, CH4 and O2 were premixed and supplied through a sintered plate into the reactor chamber to form a stable flat pilot flame, which is used to ignite and stabilize the spray-flame [30] located in the center of the pilot flame (burner nozzle). Sheath and quench gases (compressed air) were used to stabilize the flame and to cool the reactor off gas. The precursors (lanthanum(III) acetate hydrate, cobalt(II) acetate tetrahydrate, strontium(II) acetate, Fe(II) acetate) were dissolved in propionic acid and 1-propanol mixture at 60 °C, and the solution was kept at this temperature during the synthesis. The solution was transferred to two syringe pumps that were connected to a capillary (0.4 mm inner diameter and 0.7 mm outer diameter) in the center of the burner nozzle. Spray formation was achieved by atomization with the dispersion gas (O2) fed through an annular gap surrounding the capillary. The pressure inside the reactor chamber, volume flows of dispersion O2, sheath and quench gases were then adjusted to stabilize the spray-flame and reactor gas flows. During the syntheses, a filter sheet inside the filter house connected to the reactor chamber was used to filter particles from the particle-laden exhaust gas. After the synthesis, the filter house was kept under low pressure to remove physisorbed water. Then, the filter house was dismantled, and nanoparticles were harvested from the filter sheet. Table S1 shows the process parameters used for the syntheses.
Catalyst characterization
X-ray diffraction (XRD) patterns were measured with a PANalytical X’Pert PRO X-ray diffractometer operated at 40 kV and 40 mA using Ni-filtered Cu-Kα radiation and a linear position sensitive X’Celerator detector in combination with a monochromator. A silicon single crystal was used as sample holder, and the diffraction data of as-prepared nanoparticles were measured in 2θ from 15–65° with a step size of 0.03° and a scan speed of 0.05° min–1.
The whole profile fitting of XRD patterns was performed using GSASII [31]. Microstrain values obtained from Williamson–Hall plots in our previous study [32] were used and not further refined. For the Fe20, the lattice strain of 0.0047 was calculated from the Williamson–Hall plot based on the peak broadening of its XRD pattern. Polynomial background function, scale factor, lattice parameters and crystallite sizes were refined, and the results are tabulated in Table S2.
Raman spectra were measured using a He:Ne Laser with 633 nm and with a Power of ~ 0.85 mW. Each spectrum was obtained with 100 scans, and the measurement spot size is ~ 15 μm. The Raman signals were calibrated with respect to the reference signals of a silicon wafer around 520 nm.
High-angle annular dark field scanning transmission electron microscopy HAADF-STEM), and energy-dispersive X-ray spectroscopy (EDX) were performed at a JEM-2200FS (JEOL, Akishima, Japan). As-prepared nanoparticles were ultrasonically dispersed in ethanol for 5 min, and a droplet from the dispersion was dropped on a carbon-coated copper grid for TEM measurements.
Simultaneous thermal analysis (STA) measurements coupled with mass spectrometry (QMS) were performed on a NETZSCH STA 449F1 with a built-in QMS. Approximately 40 mg of the powder samples in Al2O3 crucibles were heated between 37 and 1200 ºC with a heating rate of 10 K min−1 under Ar.
A VersaProbe II (Ulvac-Phi, Chanhassen, USA) was used to measure XPS spectra of the nanoparticles. Al Kα light (\(h\nu\)= 1487 eV) with a pass energy of 12.3 eV was used to obtain the regional XPS spectra of Co 2p, La 3d, Sr 3d, C 1 s, and O 1 s. For the measurement of Fe2p and Co2p in the Fe20 sample, Mg Kα light (\(h\nu\)= 1254 eV) was used to measure the atomic ratio of Fe/(Fe + Co). A Shirley background function was used to fit the background of all the spectra. The binding energies of all spectra were corrected to C1s at 284.8 eV. The deconvolution of O1s and Sr3d spectra was carried out referring to previous reports [33,34,35]. The deconvolution of Co2p peaks was done with respect to the detected XPS signals.
XAS data were collected at the SAMBA beamline of the SOLEIL synchrotron. The beamline uses a Si (220) double crystal monochromator for selection of the incident beam energy. The data were collected at room temperature in transmission mode using gas ionization chambers. Samples were prepared as pellets by mixing the nanoparticles with boron nitride to approximately 1% Co concentration and covered with 38 μm Kapton tape. The incident energy was calibrated by setting the first inflection point of the Co K-edge XAS spectra to 7709.0 eV for a Co foil. Data were collected at room temperature. XANES and EXAFS data were processed with the Athena program and analyzed using Artemis with the IFEFFIT software package [36]. The baseline correction of the pre-edge was done using the XAS viewer from the Larch package [37].
Electrochemical tests
The electrochemical measurements were performed in a three-electrode cell set-up using an Autolab PGSTAT bipotentiostat/galvanostat (Metrohm). Catalyst inks (5 mg mL−1) were prepared by dispersing the catalyst powder in volumetric mixtures of ultrapure water, ethanol, and Nafion® solution (~ 5 wt%, ~ 0.9 g cm−3) in the ratio of 49:49:2. After 15 min sonication, the ink was drop cast on polished glassy carbon rotating disk electrodes to obtain a catalyst mass loading of 210 µg cm−2. The catalyst-coated electrode, a platinum mesh, and an Ag/AgCl (3 M KCl) electrode were used as working, counter, and reference electrodes. The counter electrode was separated from the bulk electrolyte using a separated compartment equipped with a glass frit.
A 1 M KOH solution was purified over Chelex® 100 and used as electrolyte. To determine the uncompensated resistance (RU), electrochemical impedance spectra were collected at the open-circuit potential within a frequency range from 100 kHz to 100 Hz and using a 10 mV (RMS) AC amplitude. Prior to the activity measurements, a conditioning step was conducted by ten cyclic voltammograms in a potential window of 0.0 to 0.40 V vs. Ag/AgCl (3 M KCl) with a scan rate of 100 mV/s without electrode rotation. Then, activity measurements were done by linear sweep voltammetry in the potential window of 0.0 to 0.8 V vs. Ag/AgCl (3 M KCl) with a scan rate of 5 mV s−1 at a rotation speed of 1600 rpm. All experiments were performed three times for each catalyst. The OER stability was determined from chronopotentiometric measurements after collection of the impedance spectra and the conditioning step as described above. The modified electrodes were held at 10 mA cm−2 for two hours at a rotation speed of 1600 rpm. The recorded potentials were converted from the Ag/AgCl (3 M KCl) scale to the reversible hydrogen scale (RHE) using Eqs. 1 and 2.
ERHE is the working electrode potential with reference to RHE, Emeasured is the measured potential at the working electrode with reference to Ag/AgCl (3 M KCl), E0Ag/AgCl (3 M KCl) is the formal potential of Ag/AgCl (3 M KCl) vs. RHE, pH denotes the pH value, which was obtained considering the OH– concentration and using an average value of γ = 0.766 [38,39,40] for the activity coefficient of KOH in water (Eq. 2), i is the measured current and Ru the uncompensated resistance.
The electrochemical active surface area (ECSA) for each catalyst was estimated from the electrochemical double-layer capacitance (CDL). Cyclic voltammograms were recorded in the OCP ± 0.05 V potential window using different scan rates (ν): 0.005, 0.01, 0.02, 0.025, 0.03, 0.04 and 0.05 V/s. 10 s waiting time was used between the anodic and the cathodic scans. The measurements were performed triple for each catalyst. The double-layer charging current iC was plotted against the scan rate (ν), and the electrochemical double-layer capacitance was obtained by extracting the slope of the resulting line using Eq. (3). The charging current iC was calculated from the average of the absolute currents from the anodic and the cathodic scan of the CV for a given potential in the non-Faradaic region at the respective scan rate.
The corresponding CDL value was used to calculate the ECSA, using Eq. (4).
where CS is the specific capacitance in alkaline solution, which is approximately 0.04 mF cm−2 [41].
Results and discussion
Catalyst characterization
Pristine, Fe-/Sr-substituted LaCoO3 nanoparticles were synthesized by spray-flame synthesis and characterized in the as-prepared state. LaCoO3 nanoparticles were labeled as LCO, while the molar fractions of Sr and Fe precursors were used for denoting the substituted LCO; i.e., Sr- and Fe-containing perovskites were labeled as Sr10, Sr20, and Fe20.
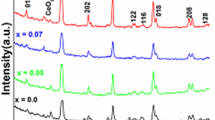
The phase composition of as-prepared nanoparticles was determined by XRD measurements shown in Fig. 1. The crystalline nature was identified by the intense and narrow diffraction peaks and confirmed by STEM measurements showing highly crystalline perovskite crystals (Fig. S2a). The surface morphology of all the perovskite crystallites shows a similar crystalline structure, and no notable change is observed in the surface structure of LCO after Fe- or Sr- substitution (the inset in Fig. S2a). All XRD patterns predominantly match with those of the rhombohedrally distorted cubic structure with the space group of R \(\overline{3 }\) c [42]. It is noteworthy to mention that the observed XRD patterns can also have relevance to the diffraction peaks of oxygen-deficient LaCoO2.937 with the space group of R \(\overline{3 }\) c H (ICSD: 153995) as stated in our previous report [32]. The most intense diffraction peak of LCO at 2θ ~ 32.8° and the broad peak at 2θ ~ 47.3° show an increase upon Sr substitution while decreasing upon Fe substitution (Fig. S2b). To clarify the changes in the peak shapes, the whole pattern refinements were done, and the lattice parameters and crystallite sizes were calculated (Table S2). Accordingly, the lattice parameters of LCO in the a and c axis show a marginal increase with Sr substitution. With Fe substitution, the lattice parameter of LCO in the a axis increases pronouncedly, while that in the c axis shows a notable decrease. These results suggest that compared to Sr substitution, Fe substitution into LCO creates a significant change in their lattice constants. In addition, the crystallite sizes calculated from pure and substituted LCO show an increasing trend in the order: Fe20 < LCO < Sr10 < Sr20. The crystallite size order is mainly in agreement with the TEM particle sizes of the samples as indicated in Table S2. These results also suggest that the differences in XRD peak intensities of Sr10, Sr20, Fe20, and LCO at 2θ ~ 32.8° and ~ 47.3° are most likely originating from the differences in the peak broadenings. It should be noted that such broadening can be not only due to different crystallite sizes but also to lower microstrains of Sr10 and Sr20 than in LCO as calculated from Williamson–Hall analysis in our previous report [32].

XRD patterns of as-prepared pristine, and Fe-/Sr-substituted LaCoO3 perovskite nanoparticles
The inset in Fig. 1 gives more insight into the crystal structures of the perovskite samples. Sr substitution results in higher diffraction peaks at 2θ ~ 46.8°, shown by the black arrow, next to the main diffraction peak of R \(\overline{3 }\) c at 2θ ~ 47.3°. These findings indicate the presence of another crystal structure or local structuring in Sr-substituted LCO crystallites. Thus, the crystalline symmetry of the perovskites was further investigated using Raman spectroscopy measurements to explain the observed changes in the XRD patterns, and the results are shown in Fig. 2.

Normalized Raman spectra of the pristine and Fe-/Sr-substituted LaCoO3 perovskite nanoparticles
The Raman spectrum of LCO shows sharp bands at ~ 147 and 678 cm–1 together with the minor bands at 400, 472, 608 cm–1. The band at ~ 147 cm–1 is associated with Eg La stretching vibration, while that at ~ 678 cm–1 corresponds to the breathing A2g mode of oxygen ion cage [43]. Although the band at ~ 678 cm–1 is Raman-inactive for rhombohedral perovskites, they show high scattering intensity due to strong electron–phonon interactions [43]. These bands can be associated with breathing A2g mode in monoclinic I2/a symmetry [44]. Alternatively, these bands can be also related to local Jahn–Teller (JT) distortions in the semiconducting phases of cobaltite perovskites [45]. The band at ~ 400 cm–1 can be associated with intermediate spin state (IS) in LaCoO3 [44]. The band at ~ 472 cm–1 can be related to the Eg bending vibrations in the non-JT distorted phase [45]. The broad band between ~ 550 and 650 cm–1 shows a peak at ~ 608 cm–1. This band can be assigned to the bending-type vibrations of Ag and Bg symmetry in the JT distorted phase.
Upon Fe substitution, broadening of the band at ~ 678 cm–1 toward lower wavenumbers is visible, and no splitting of the bands at ~ 608 cm–1 appears. Higher broadening and smaller splitting of phonon bands were previously detected in infrared reflectivity spectra of LaFeO3 compared to that of LaCoO3 [46]. These findings suggest the broadened bands between ~ 600 and 670 cm–1 may be associated with orthorhombic distortions in Fe20, as these distortions are mainly related to rigid rotation of the octahedra, and thus, have little influence on the splitting of phonon bands [46]. Furthermore, the band intensity at ~ 400 cm–1 decreases significantly, and this finding can be attributed to a lower content of IS states in Fe20 compared to LCO. Besides, the multi-phonon scattering bands are visible at ~ 343 and 1586 cm–1 in Fe20 and they likely appear due to much higher Raman signals detected for Fe20 compared to other samples (i.e., approximately six times more intensity in Fe20).
Upon Sr substitution, the scattering intensity of the bands at ~ 147 cm–1 appears higher relative to those between ~ 450 and 800 cm–1. Higher intensities at ~ 147 cm–1 correspond to the increasing Eg stretching vibration of La upon Sr substitution. Compared to LCO, Sr10 and Sr20 do not show intense features at 608 cm−1, and this finding can be associated with lower degree of JT distortion in Sr-substituted LaCoO3.
Monoclinic distortions in the crystal structure of cobaltite perovskites were previously associated with the presence of the intermediate spin state of Co3+ ions [47]. Thus, for a more detailed analysis, the electronic structure of Fe-/Sr-substituted LCO systems was further investigated using X-ray absorption spectroscopy (XAS).
The Co K-edge XAS spectra of powder samples measured in transmission mode are shown in Fig. 3. The spectra are fairly similar with some subtle differences in the pre-edge and white line regions. The pre-edge features originate from the quadrupole-allowed, dipole-forbidden 1s→3d transitions. For the Co3+ ion in these perovskites, the 3d levels are anti-bonding t2g* and eg* orbitals. The lowest energy configuration expected for the Co3+ is a low-spin (LS) (t2g6) configuration, which should have only one pre-edge feature (from 1 s → eg*). Nevertheless, intermediate-state (IS) (t2g5 eg1) and high-spin (HS) (t2g4 eg2) configurations have been shown to be accessible at room temperature [48]. The pre-edge of the LCO shows two features separated by 2 eV, indicating that the Co ions are partially in the IS and/or HS configuration. In addition to the transition to the partially filled t2g orbitals from the HS and IS configurations, there are several additional mechanisms that would explain the intensity of the low energy feature (7709.5 eV) of the pre-edge. Based on the analysis of XPS satellite features in Fig. S3, Co2p spectra show that the Co2+ content in Sr10, Sr20 and LCO is slightly higher than in Fe20, and this more reduced fraction would contribute to the low energy region of the pre-edge. Additionally, it has been described that for cobaltite perovskites, ligand to metal charge transfer (LMCT) plays an important role, increasing the amount of ligand holes and oxygen vacancies [49, 50].

a Normalized Co K-edge XAS of the pristine and Fe-/Sr-substituted LCO. The inset shows the pre-edge region in detail. b The magnitude part of the Fourier transform of the EXAFS
The LCO has the highest intensity in the pre-edge region, which could be explained by a higher oxidation state (more d-holes), a decreased separation between the t2g and eg orbitals or higher JT distortion. The position of the rising main edge (corresponding to 1 s → 4p transitions) is also sensitive to the oxidation state of the absorbing ion. In Fig. 3, the position of the edge does not significantly change among the samples. When determined from the first derivative maximum, the LCO edge is slightly shifted to higher energy (~ 0.1 eV), which is in agreement with the pre-edge intensity and position and indicates a small decrease in valency for the Sr- or Fe- containing perovskites. A previous report by Haas et al. regarding a series of Sr-substituted lanthanum cobaltite perovskites (xSr = 0.3 and 0.5) showed a decrease in the high-energy feature of the pre-edge upon Sr loading, which the authors attributed to increased LMCT contributions upon Sr loading as a result of oxygen vacancies [49].
Another interesting feature of the XAS spectra is the decrease in the maximum intensity at the absorption edge (white line), also observed by Haas et al. [49], and assigned to a decrease in covalency, LMCT-induced shake down processes, and increase in the disorder in the structure. The introduction of Sr induces distortion and disorder, which results blurring the absorption process along the energy axis and therefore lowering the intensity of the white line. We can see that the effect is absent for the Fe-substituted perovskite, since the Fe3+ ion does not induce as much distortion at the Co. The extended X-ray absorption fine structures (EXAFS) also support this point. Figure 3b shows the Fourier transform (FT) of the k3-weighted EXAFS data. These spectra indicate that the overall structure of the cobaltite perovskite is maintained upon substitution with Sr or Fe. The most intense peak R = 1.5 Å corresponds to the Co–O scattering path, while the rest of the features arise from several single and multiple scattering paths, e.g., Co–La, Co–Co–Sr, Co–O–O, Co–O–Co. Additionally, one should also consider that the samples contain mixtures of rhombohedrally distorted cubic and monoclinic structures. As a result, the EXAFS fitting and interpretation of the full R-space is very complex. Nevertheless, the first shell Co–O peak can be accurately fitted. We used this fitting to obtain accurate Co–O distances, which can be related to the valence state of the Co, as well as the coordination number to interrogate oxygen vacancies. Although the different perovskite structures can have some Jahn–Teller distortions which results in several different distances for the Co–O, the resolution of the FT EXAFS does not allow unambiguous determination of three different Co–O distances. We did two types of fits, one where the coordination number (CN) was allowed to float and another where it was fixed to 6. For both fittings protocols, the amplitude reduction factor (\({S}_{0}^{2}\)) was set to 0.9 and the Co–O distance, edge energy correction (ΔE0) and the Debye–Waller factor (σ2) were allowed to float. The R-factor gives a fractional misfit (not scaled by the data uncertainty).
The results shown in Table S3 and Table S4 show very small variations for the Co–O distance, and almost identical values for both fittings. The shortest Co–O distance was fitted for the LCO sample, in agreement with the slightly higher oxidation state indicated by the edge position and the pre-edge intensity. The variations among the other samples are within the error limit. As mentioned above, a higher σ2 indicates a more disordered structure for the Sr10 and Sr20 perovskites, and it is also reflected by the intensity decrease in the features at R = 3 Å, indicating a long-range disorder beyond the first coordination sphere. The fitting of the coordination number indicates that the maximum oxygen vacancies are present in the Sr20 sample, while the other three have similar values (Table S3). This result is consistent with the expectation that Sr2+ substitution of La3+ ions must be balanced by ligand holes.
Besides the structural analysis of the Fe/Sr-substituted perovskites, the temperature response was also investigated by heating the as-prepared nanoparticles in a controlled atmosphere to determine organic species released from the samples. For this purpose, the samples were heated under Ar from 37 to 1200 °C, and the total weight loss and the nature of the released species were determined by STA coupled with mass spectrometry (QMS) as shown in Fig. 4a. The STA curves of all samples show a mass loss of about 4.7 wt% until 300 °C, which can mainly be attributed to the release of adsorbed H2O and CO2 originating from burning residuals as identified in the mass spectrometry graphs shown in the inset. On top, the STA curves indicated that LCO, Sr10, and Sr20 have higher mass losses between 300 and 620 °C compared to Fe20 (~ 3.0 wt% vs. 1.3 wt%), and this result can be correlated with more intense QMS signals for H2O and CO2 as demonstrated in the inset and in Fig. 4b. The sharp signals at ~ 325 °C can be associated with H2O desorption of surface-bound La(OH)3 while those at 380–390 °C can be related to CO2 desorption of surface-bound SrCO3 and La2O2CO3 [51]. The mass losses of LCO, Sr10 and Sr20 between 500 and 800 °C correlate with increasing QMS signals of CO2. Signals at ~ 700 °C were previously observed for the CO2-TPD profiles of Sr-, Co-, and Fe-based perovskites [52], and they suggest the thermal decomposition of bulk CO3 species, i.e., SrCO3 and La2O2CO3.

a STA measurement of pristine and Fe-/Sr-substituted LaCoO3 and QMS measurement of H2O (inset). b QMS measurement of CO2 released from the same measurement
Overall, Fe-substituted LaCoO3 perovskites mostly impede incorporation of carbonate ions into perovskites and their mass loss is mainly due to the release of adsorbed water and CO2 from burning residuals. On the other hand, pristine and Sr-substituted LaCoO3 perovskites have much higher mass losses due to the bulk CO2 species decomposed from SrCO3 and La2O2CO3 at high temperatures.
While the B-site (Fe vs. Co)-substituted LaCoO3 perovskites typically show homogeneous substitutional doping, A-site-substituted La1–xSrxCoO3 perovskite crystals often lack from an inhomogeneous distribution of Sr atoms with excess Sr at the surface [18]. Thus, the materials were investigated by XPS measurements. Mg-Kα radiation (E = 1254 eV) was used to eliminate the Auger peaks of cobalt on Fe2p spectra for the quantification of Fe and Co XPS signals. The integral areas of the La3d, Sr3d, Co2p and Fe2p spectra were calculated, and the results are summarized in Table 1. In case of Fe20 (Fig. S4), the ratio Fe/(Fe + Co) indicating the degree of Fe substitution was found near 0.21, being close to the intended ratio of 0.25, and indicating homogeneous distribution of Fe atoms in Fe20. La/(Co + Fe) of 0.95 and O/(Co + Fe) atom ratios of 2.93 also confirm that, the surface composition of Fe20 is close to the stoichiometric perovskite composition. In LCO, the La/Co ratio of 0.95 is similar to those of Fe20, but an O/Co ratio of about 2.78 suggests a higher surface oxygen deficiency in this sample, in agreement with the EXAFS fittings shown above.
For Sr10 and Sr20, SrCO3 could be identified from the regional XPS Sr3d spectra (see Fig. S5) as also assumed from the STA analysis. For the compositional analysis, the integral area of Sr originating from SrCO3 was subtracted from the integral area of the Sr3d spectra to determine the Sr atoms associated with perovskites (see Sr/(Sr + La) and (La + Sr)/Co in the parentheses). Sr/(Sr + La) ratios corresponding to 20 and 33 at% of Sr were obtained for Sr10 and Sr20 and confirm the excess amount of Sr atoms. The (La + Sr)/Co ratio of 0.92–0.97 and O/Co of 2.80 in Sr10 is close to those of LCO and Fe20. In Sr20, the A-site surface atoms are slightly more concentrated, and the reduction in the (La + Sr)/Co ratio near 1.00 (after subtracting the SrCO3 signals from Sr3d spectra) implies an excess of A-site atoms due to the formation of SrCO3. Also, the surface O/Co ratio of Sr20 was higher than for the other samples, in contrast to what was observed in EXAFS. This excess oxygen can also be assigned to surface SrCO3 and/or Sr(OH)2 species. To summarize, Sr atoms are enriched at the surface of Sr10 and Sr20, which can be primarily attributed to the surface formation of SrCO3. This effect is more pronounced at higher Sr substitutions.
Segregation into multiple phases is also observed when heating as-synthesized Sr20 for 5 h at 250 °C under oxygen. The comparison of its diffraction pattern with that of as-prepared Sr20 shows that after the heat-treatment SrCO3, Co3O4 and La2CoO4 phases appear (Fig. S6). These results suggest some metastability of the as-prepared material, which is supported by the findings from EXAFS which demonstrated a long-range disorder beyond the first coordination sphere, especially for Sr20.
Next to the physical characterization of the as-prepared perovskite samples, their OER catalytic activities were investigated by linear sweep voltammetry using the rotating disk electrode technique. The recorded polarization curves normalized to the geometric and the determined electrochemical surface area (ECSA) are shown in Fig. 5a and b, respectively. Partial substitution of the LaCoO3 perovskite with Fe or Sr has an evident influence on the OER activity. While LCO requires 400 mV overpotential to produce O2 with a current density of 10 mA cm−2, lower overpotentials of 0.35, 0.34, and 0.37 V are required for Sr10, Sr20, and Fe20, respectively. Sr20 shows the highest OER activity, followed by Sr10 and Fe20. Still, the differences between the three samples are relatively small. The ECSA, calculated based on the capacitive currents recorded with different scan rates (Fig. S7), highlights an increased electrochemically active surface area for Sr20 followed by Sr10, Fe20, and LCO. Therefore, normalization of the current recorded during the linear sweep voltammetry (LSV) was performed. The data are shown in Fig. 5b. However, in this case, too, the substituted samples show an increased activity compared with LCO. Sr20 again offers the highest activity, but this time, the differences between Sr10, Sr20 and Fe20 are less pronounced. It is here noteworthy that the OER activity of Sr20 is comparable to or superior to the other OER catalysts based on cobaltite perovskite structures in the literature (Table S5). The stability of Sr10 and Sr20 catalysts was evaluated by chronopotentiometry performed at 10 mA cm−2 for 2 h (Fig. 5c). For Sr10 and S20, a constant potential is recorded during the 2 h galvanostatic electrolysis conducted at a current density of 10 mA cm−2. Two measurements were performed for the two catalysts to confirm the results. During the 2 h electrolysis, no significant increase in the potential is recorded, indicating that the catalysts show a stable electrocatalytic performance at moderate conditions.

Comparison of the electrocatalytic OER activity of pristine and Fe-/Sr-substituted LaCoO3 catalysts in 1 M KOH at a scan rate of 5 mV s−1 and 1600 rpm. Current densities are calculated by normalization of the recorded currents to a the geometric area of the electrode or b the electrochemical active surface area (ECSA) calculated from double-layer capacitance. c Electrocatalytic OER stability of Sr10 and Sr20 catalysts determined by 2 h chronopotentiometric measurements at 10 mA cm−2 in 1 M KOH at 1600 rpm
To be able to relate the OER activity trends with the analyzed material properties of the catalysts, the characterization results in this study and our previous study upon Sr-substituted LaCoO3 [32] are summarized in Table S6. Accordingly, the ECSA values directly correlate with the OER activity trends and thus, can be considered the major factor affecting the OER activity of Sr20, Sr10 and Fe20. After normalizing the recorded currents to the ECSA, Sr20 still had a slightly higher OER activity than the other substituted catalysts. This could be related to the combined effect of the lower coordination number of Co–O and the more disordered structure (i.e., relatively high Debye–Waller factor) in Sr20. The lower coordination number of Co–O in Sr20 can originate from a relatively higher oxygen vacancy concentration as well as a more disordered and complex perovskite structure allowing several Co–O distances. To illustrate such structures, the atomic structure of a single crystallite of 30 at. % Sr-substituted LaCoO3 was already shown in our previous study to demonstrate the oxygen vacancy ordering and disordered atomic structure in Sr-substituted perovskites [32]. On the other hand, LCO clearly had a lower OER activity than the other catalysts and this could be due to its lower structural stability under OER conditions owing to the combined effect of high microstrain and low crystallite size. The other changes in the bulk and surface properties of these catalysts in Table S5 do not show a direct correlation with their OER activity trend.
Conclusions
Spray-flame synthesis was successfully applied to produce phase-pure pristine, Fe- and Sr-substituted LaCoO3 perovskite nanoparticles as potential OER catalyst materials. The as-prepared nanoparticles have a crystalline structure and essentially belong to the rhombohedrally distorted cubic structure. Raman and XPS spectra analyses suggest that A-site substitution by Sr leads to a lower JT effect in the perovskite phase while having comparable surface Co2+ concentration with pristine LCO. On the other hand, B-site substitution by Fe results in an orthorhombic distortion and a lower content of IS states in the perovskite phase, while increasing the concentration of surface Co3+. XAS analyses indicate that the perovskite structure of LaCoO3 is maintained upon Fe and Sr substitution. EXAFS fittings of the first shell of Co–O peaks show that substitutional doping with Sr induces a more disordered structure together with long-range disorder. Fittings of the coordination number indicate a lower Co–O coordination number in Sr20 compared to other samples. Fe substitution does not result in considerable changes of the perovskite structure. Simultaneous thermal analysis (STA) coupled with mass spectrometry measurements reveals higher mass losses occurring in Sr-substituted La1–xSrxCoO3 compared to the other perovskites. This finding can be associated with the thermal decomposition of these compounds into other perovskite-like structures, formation of cobalt spinel phases and decomposition of carbonates. The results from survey XPS measurements point out near-stoichiometric surface compositions of the catalysts except for La0.8Sr0.2CoO3 on which Sr segregation was found. In accordance with the STA results, the nature of the Sr segregated groups in as-prepared particles could be attributed to Sr carbonates on perovskite crystals. The catalytic activity measurements of the perovskite nanoparticles indicate a higher activity of the Fe-/Sr-substituted perovskites compared to phase pure LCO. The 20 at% Sr-substituted catalyst shows the highest OER activity with an overpotential of only 0.34 V, and a good catalytic stability at least for 2 h under a current density of 10 mA cm−2. The higher activity of Sr20 could be attributed to its higher ECSA and is possibly influenced by a lower coordination number in Co–O as well as a higher degree of long-range disorder.
References
Schmidt O, Gambhir A, Staffell I et al (2017) Future cost and performance of water electrolysis: an expert elicitation study. Int J Hydrogen Energy 42:30470–30492. https://doi.org/10.1016/j.ijhydene.2017.10.045
Buttler A, Spliethoff H (2018) Current status of water electrolysis for energy storage, grid balancing and sector coupling via power-to-gas and power-to-liquids: a review. Renew Sustain Energy Rev 82:2440–2454. https://doi.org/10.1016/j.rser.2017.09.003
Suen N-T, Hung S-F, Quan Q et al (2017) Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives. Chem Soc Rev 46:337–365. https://doi.org/10.1039/C6CS00328A
Dau H, Limberg C, Reier T et al (2010) The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis. ChemCatChem 2:724–761. https://doi.org/10.1002/cctc.201000126
Li Y, Du X, Huang J et al (2019) Recent progress on surface reconstruction of earth-abundant electrocatalysts for water oxidation. Small 15:1901980. https://doi.org/10.1002/smll.201901980
Song F, Bai L, Moysiadou A et al (2018) Transition metal oxides as electrocatalysts for the oxygen evolution reaction in alkaline solutions: an application-inspired renaissance. J Am Chem Soc 140:7748–7759. https://doi.org/10.1021/jacs.8b04546
Man IC, Su HY, Calle-Vallejo F et al (2011) Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3:1159–1165. https://doi.org/10.1002/cctc.201000397
Zhu H, Zhang P, Dai S (2015) Recent advances of lanthanum-based perovskite oxides for catalysis. ACS Catal 5:6370–6385. https://doi.org/10.1021/acscatal.5b01667
Hwang J, Rao RR, Giordano L et al (2017) Perovskites in catalysis and electrocatalysis. Science 358:751–756. https://doi.org/10.1126/science.aam7092
Yildiz B (2014) Stretching the energy landscape of oxides—effects on electrocatalysis and diffusion. MRS Bull 39:147–156. https://doi.org/10.1557/mrs.2014.8
Riva M, Kubicek M, Hao X et al (2018) Influence of surface atomic structure demonstrated on oxygen incorporation mechanism at a model perovskite oxide. Nat Commun 9:3710. https://doi.org/10.1038/s41467-018-05685-5
Weng B, Song Z, Zhu R et al (2020) Simple descriptor derived from symbolic regression accelerating the discovery of new perovskite catalysts. Nat Commun 11:3513. https://doi.org/10.1038/s41467-020-17263-9
Palkovits R, Palkovits S (2019) Using artificial intelligence to forecast water oxidation catalysts. ACS Catal 9:8383–8387. https://doi.org/10.1021/acscatal.9b01985
Tong Y, Guo Y, Chen P et al (2017) Spin-state regulation of perovskite cobaltite to realize enhanced oxygen evolution activity. Chem 3:812–821. https://doi.org/10.1016/j.chempr.2017.09.003
Yun TG, Heo Y, Bin Bae H, Chung SY (2021) Elucidating intrinsic contribution of d-orbital states to oxygen evolution electrocatalysis in oxides. Nat Commun 12:824. https://doi.org/10.1038/s41467-021-21055-0
Sun C, Alonso JA, Bian J (2021) Recent advances in perovskite-type oxides for energy conversion and storage applications. Adv Energy Mater 11:2000459. https://doi.org/10.1002/aenm.202000459
Mefford JT, Rong X, Abakumov AM et al (2016) Water electrolysis on La1–xSrxCoO3–δ perovskite electrocatalysts. Nat Commun 7:11053. https://doi.org/10.1038/ncomms11053
Koo B, Kim K, Kim JK et al (2018) Sr segregation in perovskite oxides: Why it happens and how it exists. Joule 2:1476–1499. https://doi.org/10.1016/j.joule.2018.07.016
Wang Y, Shen X, Arandiyan H et al (2020) Tuning the surface energy density of non-stoichiometric LaCoO3 perovskite for enhanced water oxidation. J Power Sources 478:228748. https://doi.org/10.1016/j.jpowsour.2020.228748
Mueller DN, Giesen M, Duchoň T et al (2021) Nanoscopic surface decomposition of Pr0.5Ba0.5CoO3−δ perovskites turns performance descriptors ambiguous. J Phys Chem 125:10043–10050. https://doi.org/10.1021/acs.jpcc.1c00976
Sun J, Du L, Sun B et al (2020) Bifunctional LaMn0.3Co0.7O3 perovskite oxide catalyst for oxygen reduction and evolution reactions: the optimized eg electronic structures by manganese dopant. ACS Appl Mater Interfaces 12:24717–24725. https://doi.org/10.1021/acsami.0c03983
Duan Y, Sun S, Xi S et al (2017) Tailoring the Co 3d-O 2p covalency in LaCoO3 by Fe substitution to promote oxygen evolution reaction. Chem Mater 29:10534–10541. https://doi.org/10.1021/acs.chemmater.7b04534
Mohan S, Mao Y (2020) Molten salt synthesized submicron perovskite La1–xSrxCoO3 particles as efficient electrocatalyst for water electrolysis. Front Mater 7:259. https://doi.org/10.3389/fmats.2020.00259
Alkan B, Cychy S, Varhade S et al (2019) Spray-flame-synthesized LaCo1–xFexO3 perovskite nanoparticles as electrocatalysts for water and ethanol oxidation. ChemElectroChem 6:4266–4274. https://doi.org/10.1002/celc.201900168
Alkan B (2020) Spray-flame-synthesized LaCo1–xFexO3±δ perovskites for electrocatalysis. PhD thesis, University of Duisburg-Essen https://doi.org/10.17185/duepublico/71937
Alkan B, Medina D, Landers J et al (2020) Spray-flame-prepared LaCo1–xFexO3 perovskite nanoparticles as active OER catalysts: influence of Fe content and low-temperature heating. ChemElectroChem 7:2564–2574. https://doi.org/10.1002/celc.201902051
Hardt S, Wlokas I, Schulz C, Wiggers H (2015) Impact of ambient pressure on titania nanoparticle formation during spray-flame synthesis. J Nanosci Nanotechnol 15:9449–9456. https://doi.org/10.1166/jnn.2015.10607
Angel S, Tapia JD, Gallego J et al (2021) Spray-flame synthesis of LaMnO3+δ nanoparticles for selective CO oxidation (SELOX). Energ Fuels 35:4367–4376. https://doi.org/10.1021/acs.energyfuels.0c03659
Tarasov A, Shvartsman VV, Shoja S et al (2020) Spray-flame synthesis of BaTi1–xZrxO3 nanoparticles for energy storage applications. Ceram Int 46:13915–13924. https://doi.org/10.1016/j.ceramint.2020.02.187
Schneider F, Suleiman S, Menser J et al (2019) SpraySyn—a standardized burner configuration for nanoparticle synthesis in spray flames. Rev Sci Instrum 90:085108. https://doi.org/10.1063/1.5090232
Toby BH, Von Dreele RB (2013) GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J Appl Crystallogr 46(2):544–549. https://doi.org/10.1107/S0021889813003531
Büker J, Alkan B, Chabbra S et al (2021) Liquid-phase cyclohexene oxidation with O2 over spray-flame-synthesized La1–xSrxCoO3 perovskite nanoparticles. Chem Eur J 27:16912–16923. https://doi.org/10.1002/chem.202103381
Knipe SW, Mycroft JR, Pratt AR et al (1995) X-ray photoelectron spectroscopic study of water adsorption on iron sulphide minerals. Geochim Cosmochim Acta 59:1079–1090. https://doi.org/10.1016/0016-7037(95)00025-U
Wang CC, Jiang SP (2015) Mechanism of SO2 poisoning on the electrochemical activity of LSCF and LSM electrodes. ECS Trans 68:1023–1029
Vasquez RP (1992) SrSO4 by XPS. Surf Sci Spectra 1:117–121. https://doi.org/10.1116/1.1247681
Ravel B, Newville M (2005) ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Radiat 12:537–541. https://doi.org/10.1107/S0909049505012719
Newville M (2013) Larch: an analysis package for XAFS and related spectroscopies. J Phys Conf Ser 430:012007. https://doi.org/10.1088/1742-6596/430/1/012007
Bromley LA (1973) Thermodynamic properties of strong electrolytes in aqueous solutions. AIChE J 19:313–320. https://doi.org/10.1002/aic.690190216
Knobel M (1923) The activities of the ions of potassium hydroxide in aqueous solution. J Am Chem Soc 45:70–76. https://doi.org/10.1021/ja01654a010
Hamer WJ, Wu YC (1972) Osmotic coefficients and mean activity coefficients of uni-univalent electrolytes in water at 25° C. J Phys Chem Ref Data 1(4):1047–1100. https://doi.org/10.1063/1.3253108
McCrory CCL, Jung S, Peters JC, Jaramillo TF (2013) Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J Am Chem Soc 135:16977–16987. https://doi.org/10.1021/ja407115p
Radaelli PG, Cheong S-W (2002) Structural phenomena associated with the spin-state transition in LaCoO3. Phys Rev B 66:094408. https://doi.org/10.1103/PhysRevB.66.094408
Hong WT, Gadre M, Lee Y et al (2013) Tuning the spin state in LaCoO3 thin films for enhanced high- temperature oxygen electrocatalysis. J Phys Chem Lett 4:2493–2499. https://doi.org/10.1021/jz401271m
Ishikawa A, Nohara J, Sugai S (2004) Raman study of the orbital-phonon coupling in LaCoO3. Phys Rev Lett 93:136401. https://doi.org/10.1103/PhysRevLett.93.136401
Orlovskaya N, Steinmetz D, Yarmolenko S et al (2005) Detection of temperature- and stress-induced modifications of LaCoO3 by micro-Raman spectroscopy. Phys Rev B Condens Matter Mater Phys 72:1–7. https://doi.org/10.1103/PhysRevB.72.014122
Tajima S, Masaki A, Uchida S et al (1987) Infrared reflectivity and electronic states in perovskite-type oxides La1–xSrxFeO3 and La1–xSrxCoO3. J Phys C Solid State Phys 20:3469–3484. https://doi.org/10.1088/0022-3719/20/23/016
Gnezdilov V, Choi KY, Paskevich Y et al (2006) Low temperature mixed spin state of Co3+ in LaCoO3 evidenced from Jahn-Teller lattice distortions. Fiz Nizk Temp 32:219–226. https://doi.org/10.1063/1.2171521
Vankó G, Rueff JP, Mattila A et al (2006) Temperature- and pressure-induced spin-state transitions in LaCoO3. Phys Rev B 73:024424. https://doi.org/10.1103/PhysRevB.73.024424
Haas O, Ludwig C, Bergmann U et al (2011) X-ray absorption investigation of the valence state and electronic structure of La1–xCaxCoO3–δ in comparison with La1–xSrxCoO3–δ and La1–xSrxFeO3–δ. J Solid State Chem 184:3163–3171. https://doi.org/10.1016/j.jssc.2011.09.027
Medarde M, Dallera C, Grioni M et al (2006) Low-temperature spin-state transition in LaCoO3 investigated using resonant x-ray absorption at the Co K edge. Phys Rev B 73:1–054424. https://doi.org/10.1103/PhysRevB.73.054424
Oemar U, Ang ML, Hee WF et al (2014) Perovskite LaxM1–xNi0.8Fe0.2O3 catalyst for steam reforming of toluene: crucial role of alkaline earth metal at low steam condition. Appl Catal B Environ 148–149:231–242. https://doi.org/10.1016/j.apcatb.2013.10.001
Yan A, Maragou V, Arico A et al (2007) Investigation of a Ba0.5Sr0.5Co0.8Fe0.2O3–δ based cathode SOFC. II. The effect of CO2 on the chemical stability. Appl Catal B Environ 76:320–327. https://doi.org/10.1016/j.apcatb.2007.06.010
Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projekt number 388390466 – TRR 247 “Heterogeneous Oxidation Catalysis in the Liquid Phase,” project C02 (40536624). We thank P. Fortugno, S. Apazeller, Md Y. Ali, and M. Heidelmann for Raman spectroscopy, STA coupled with QMS, XPS, STEM, respectively. OR and SD acknowledge funding by the Max Planck Society. XAS data were collected at the SOLEIL Synchrotron (Samba beamline, proposal number 20201215).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Handling Editor: Joshua Tong.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alkan, B., Braun, M., Landrot, G. et al. Spray-flame-synthesized Sr- and Fe-substituted LaCoO3 perovskite nanoparticles with enhanced OER activities. J Mater Sci 57, 18923–18936 (2022). https://doi.org/10.1007/s10853-022-07738-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-022-07738-z